


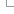
Homology types
Using the Gene Trees, we can infer the following pairwise relationships.

Orthologues
Homologues which diverged by a speciation event. There are four types of orthologues:
- 1-to-1 orthologues (ortholog_one2one)
- 1-to-many orthologues (ortholog_one2many)
- many-to-many orthologues (ortholog_many2many)
- between-species paralogues – only as exceptions
Genes in different species related by a speciation event are defined as orthologues. Depending on the number of genes found in each species, we differentiate among 1:1, 1:many and many:many relationships. Please refer to the figure, where there are examples of the three kinds.
Homoeologues
Homologues which diverged by a speciation event but end up in the same genome through hybridisation (e.g. polyploid plant genomes). They can have the same types as orthologues (see above), but importantly, the types refer to the number of genes in the sub-genomes.
Note: Homoeologues residing on a U-component (subgenome composed of unplaced assembly sequences) are treated as paralogues.
Let's take wheat (Triticum aestivum) as an example. It is a hybrid of three sub-genomes named A, B, and D. If a gene is found in one copy in every sub-genome, the three A-B, A-D, and B-D homoeologues are all labelled as 1:1 homoeologues. If the gene has a second copy in the sub-genome D, the A-B pair is labelled as 1:1, while the A-D and B-D pairs are labelled as 1:many.
Orthologues against polyploid genomes
Orthologues against a polyploid genome are labelled considering the number of genes in the sub-genomes. For example, when comparing the hexaploid wheat Triticum aestivum against barley Hordeum vulgare, a gene that is in one copy in every sub-genome and in barley will have three 1:1 orthology pairs between wheat and barley. If there are two copies in barley, there will be 3×2=6 1:many orthology pairs. If wheat's A sub-genome also has two copies of the gene, its orthology pairs with barley would be many:many, whereas the orthology pairs involving the B and D sub-genomes would still be 1:many.
Paralogues
Homologues which diverged by a duplication event. There are two types of paralogues:
- same-species paralogies (within_species_paralog)
- fragments of the same ‐predicted‐ gene (gene_split)
Genes of the same species and related by a duplication event are defined as paralogues. In the previous figure, Hsap2 and Hsap2', and Mmus2 and Mmus2' are two examples of within-species paralogues. The duplication event relating the paralogues does not need to affect this species only. For example, Mmus2' and Mmus3' are also within-species paralogues but the duplication event has occurred in the common ancestor between species Hsap (human) and species Mmus (mouse). The taxonomy level "times" the duplication event to the ancestor of Euarchontoglires.
Confidence scores
We compute a duplication confidence score for each duplication node. This is the Jaccard index of the sets of species under the two sub-trees. The score is sometimes referred to as "species intersection score".

Between-species paralogues
A between-species paralogue corresponds to a relation between genes of different species where the ancestor node has been labelled as a duplication node e.g. Mmus1:Hsap2 or Mmus1:Hsap3. Currently, we only annotate between-species paralogues when there is no better match for any of the genes, and the duplication is weakly supported (duplication confidence score < 0.25).
Such cases can be the result of real duplications followed by gene losses (as shown in the picture below), but most of the time occur as the result of a wrong gene tree topology with a spurious duplication node. Often assembly errors are behind these problems. It is not clear whether these genes are real orthologues or not, but they are the best available candidates (given the data), and we bend the definition of orthology to tag them as orthologues. They are flagged as "non-compliant with the gene tree". People interested in phylogenetic analysis mixing the orthologies and the trees should probably use the set of tree-compliant orthologies.

Gene splits
A paralogue labelled as a gene_split occurs when a gene appears to be broken in two. In the gene tree building process they will each align to one end of the other orthologues of the gene, so they will be clustered and placed in the tree alongside the other homologues. They are commonly related to fragmented genome assemblies or a gene prediction that is poor in supporting evidence, resulting in two fragmented genes where there should be a large one.
Right after building the multiple alignment, we detect pairs of genes that lie close to each other (< 1 MB) in the same sequence region and the same strand (see GeneA1/GeneA2 below). They are forced to be paired in the gene trees under gene_split events, and represent our most confident set. When there is no (or little) overlap between the gene fragments in the same species and they lie in different sequence regions in the assembly (see GeneB1/GeneB2 in the image below), we let TreeBeST [1] decide their best position in the tree. They are linked by duplication nodes, and have a lower confidence index.

Confidence scoring
We carry a number of extra analyses to assess the quality of the orthology prediction, following external sources of evidence (local synteny, and whole-genome alignments). From there we are able to flag some orthologies as high-confidence.
Notes and References
- Li, H et al., Compara-specific version, Original version (TreeBeST was previously known as NJTREE)
![[twitter logo]](/i/twitter.png "Follow us on Twitter!")
