


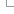
Variant Recoder
Variant Recoder is a tool for translating between different variant names. It accepts HGVS descriptions and variant identifiers from databases such as dbSNP, ClinVar, UniProt and PharmGKB (see full list of accession databases) as input.
Some forms of ambiguous and incorrect HGVS descriptions are supported including those stating only gene name and protein change (eg. AGT:p.Met259Thr) which are often seen in publications.
For each input variant, variant_recoder reports the variant identifiers held in Ensembl and HGVS genomic, transcript and protein descriptions on Ensembl, RefSeq and LRG sequences.
Web interface
The Variant Recoder web interface allows you to simply enter variant names and identifiers in a variety of formats and obtain VCF and other names/descriptions.
Input form
- First select the correct species for your data.
- You can optionally choose a name for the data you upload - this can make it easier for you to identify jobs and files that you have uploaded to the Variant Recoder at a later point.
-
You have two options for uploading your data:
- File upload - click the "Choose file" button and locate the file on your system
- Paste file - simply copy and paste the contents of your file into the large text box
The format of your data is automatically detected; the supported input data is:
We also support ambiguous descriptions listing only gene symbol and protein change (e.g. BRCA2:p.Trp31Cys), as seen in the literature. - Results: you can choose which type of data format to return. The supported formats are the same as the input plus the format VCF.
Output
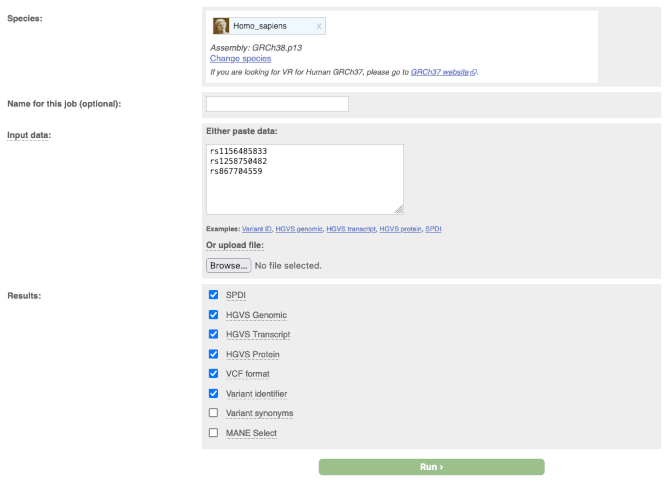
There is an output line for each alternative allele of the input variant. Each output value is annexed to the corresponding alternative allele with the exception of COSMIC and HGMD IDs.
In the output table you can click on links that will take you to known variants, where available, genomic locations or gene and transcript information.
Example of output, variant with two alternative alleles:
REST API
Ensembl provides a REST API for Variant Recoder with 2 endpoints:
- Single variant query: GET variant_recoder
- Multiple variants query: POST variant_recoder
Command line tool
Download and install
Variant Recoder is part of the Ensembl VEP package.
Please follow the instructions about the  download and installation of Ensembl VEP.
download and installation of Ensembl VEP.
Note
Usage
Variant Recoder depends on database access for identifier lookup, and cannot be used in offline mode as per Ensembl VEP.
The output format is JSON and the JSON Perl module is required.
# Running on one ID, as a string: ./variant_recoder --id [input_data_string] # Running on several IDs, in a text file: ./variant_recoder -i [input_file] --species [species]
Like Ensembl VEP, Variant Recoder can use VCF, variant identifiers, HGVS notations and SPDI in addition to the Ensembl VEP default format as input.
Output
Output is a JSON array of objects, one per input variant, with the following keys:- input: input string
- id: variant identifiers
- hgvsg: HGVS genomic nomenclature
- hgvsc: HGVS transcript nomenclature
- hgvsp: HGVS protein nomenclature
- spdi: Genomic SPDI notation
- vcf_string: VCF format (optional)
- var_synonyms: Extra known synonyms for co-located variants (optional)
- mane_select: MANE Select (Matched Annotation from NCBI and EMBL-EBI) Transcripts. (optional) Note: only available for human.
- warnings: Warnings generated e.g. for invalid HGVS
Tips
Example of output, with the --pretty flag:
./variant_recoder --id "AGT:p.Met259Thr" --pretty
[
{
"warnings" : [
"Possible invalid use of gene or protein identifier 'AGT' as HGVS reference; AGT:p.Met259Thr may resolve to multiple genomic locations"
],
"C" : {
"input" : "AGT:p.Met259Thr",
"id" : [
"rs699",
"CM920010",
"COSV64184214"
],
"hgvsg" : [
"NC_000001.11:g.230710048A>G"
],
"hgvsc" : [
"ENST00000366667.6:c.776T>C",
"ENST00000679684.1:c.776T>C",
"ENST00000679738.1:c.776T>C",
"ENST00000679802.1:c.776T>C",
"ENST00000679854.1:n.1287T>C",
"ENST00000679957.1:c.776T>C",
"ENST00000680041.1:c.776T>C",
"ENST00000680783.1:c.776T>C",
"ENST00000681269.1:c.776T>C",
"ENST00000681347.1:n.1287T>C",
"ENST00000681514.1:c.776T>C",
"ENST00000681772.1:c.776T>C",
"NM_001382817.3:c.776T>C",
"NM_001384479.1:c.776T>C"
],
"hgvsp" : [
"ENSP00000355627.5:p.Met259Thr",
"ENSP00000505981.1:p.Met259Thr",
"ENSP00000505063.1:p.Met259Thr",
"ENSP00000505184.1:p.Met259Thr",
"ENSP00000506646.1:p.Met259Thr",
"ENSP00000504866.1:p.Met259Thr",
"ENSP00000506329.1:p.Met259Thr",
"ENSP00000505985.1:p.Met259Thr",
"ENSP00000505963.1:p.Met259Thr",
"ENSP00000505829.1:p.Met259Thr",
"NP_001369746.2:p.Met259Thr",
"NP_001371408.1:p.Met259Thr"
],
"spdi" : [
"NC_000001.11:230710047:A:G"
],
}
}
]
Options
Variant Recoder shares many of the same command line flags as Ensembl VEP.
However some other flags are unique to variant_recoder:
| Flag | Alternate | Description | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
--input_data [input_string] |
-id |
A single variant as a string, such as:
|
|||||||||||||||
--input_file [input_file] |
-i |
Input file containing one or more variants, one per line. Mixed formats is disallowed.
|
|||||||||||||||
--species |
Species to use. Default value: homo_sapiens | ||||||||||||||||
--grch37 |
Use GRCh37 assembly instead of GRCh38 | ||||||||||||||||
--genomes |
Set database parameters for Ensembl Genomes species | ||||||||||||||||
--pretty |
Write pre-formatted indented JSON | ||||||||||||||||
--vcf_string |
Returns the VCF format in a string | ||||||||||||||||
--var_synonyms |
Extra known synonyms for co-located variants | ||||||||||||||||
--mane_select |
Returns MANE Select transcripts in HGVS format (e.g. hgvsg, hgvsc, hgvsp). Only available for human. | ||||||||||||||||
--fields [field1,field2] |
Limit the default output fields. Comma-separated list, one or more of: id, hgvsg, hgvsc, hgvsp, spdi. e.g.: ./variant_recoder --id "AGT:p.Met259Thr" --fields id,hgvsc |
||||||||||||||||
--host [db_host] |
Change database host from default ensembldb.ensembl.org (UK). Geographic mirrors are useastdb.ensembl.org (US East Coast) and asiadb.ensembl.org (Asia). Other flags such as --user, --port and --pass may also be set. |
||||||||||||||||
--pick, --per_gene, --pick_allele, --pick_allele_gene, --pick_order |
Set and customise transcript selection process, see Ensembl VEP documentation |
![[twitter logo]](/i/twitter.png "Follow us on Twitter!")
